
How Technology is Revolutionizing Drug Discovery: An Introduction to Computer-Aided Drug Design and Discovery


The journey of discovering and developing new drugs is known for being lengthy, risky, and expensive. On average, it takes around 14 years to bring a drug from concept to market, with costs ranging between $800 million to $1 billion.
However, advancements in combinatorial chemistry and high-throughput screening technologies have significantly sped up this process, allowing vast libraries of compounds to be quickly screened and synthesized. Even though there's been a big increase in spending on new drug development over the past few decades, the results haven't matched the investment due to the challenges of low efficiency and high failure rates in drug discovery. To tackle this, various strategies have been created to speed up research and cut costs and risks. One of the most effective ways to achieve this is through computer-aided drug design (CADD).
Computer-aided drug design (CADD) uses computer techniques to help find, develop, and study drugs and other molecules with similar biochemical properties. Using computer simulations, undesirable compounds with poor activity or unfavorable ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties can be filtered out, leaving the most promising candidates.
Stages Of Drug Discovery
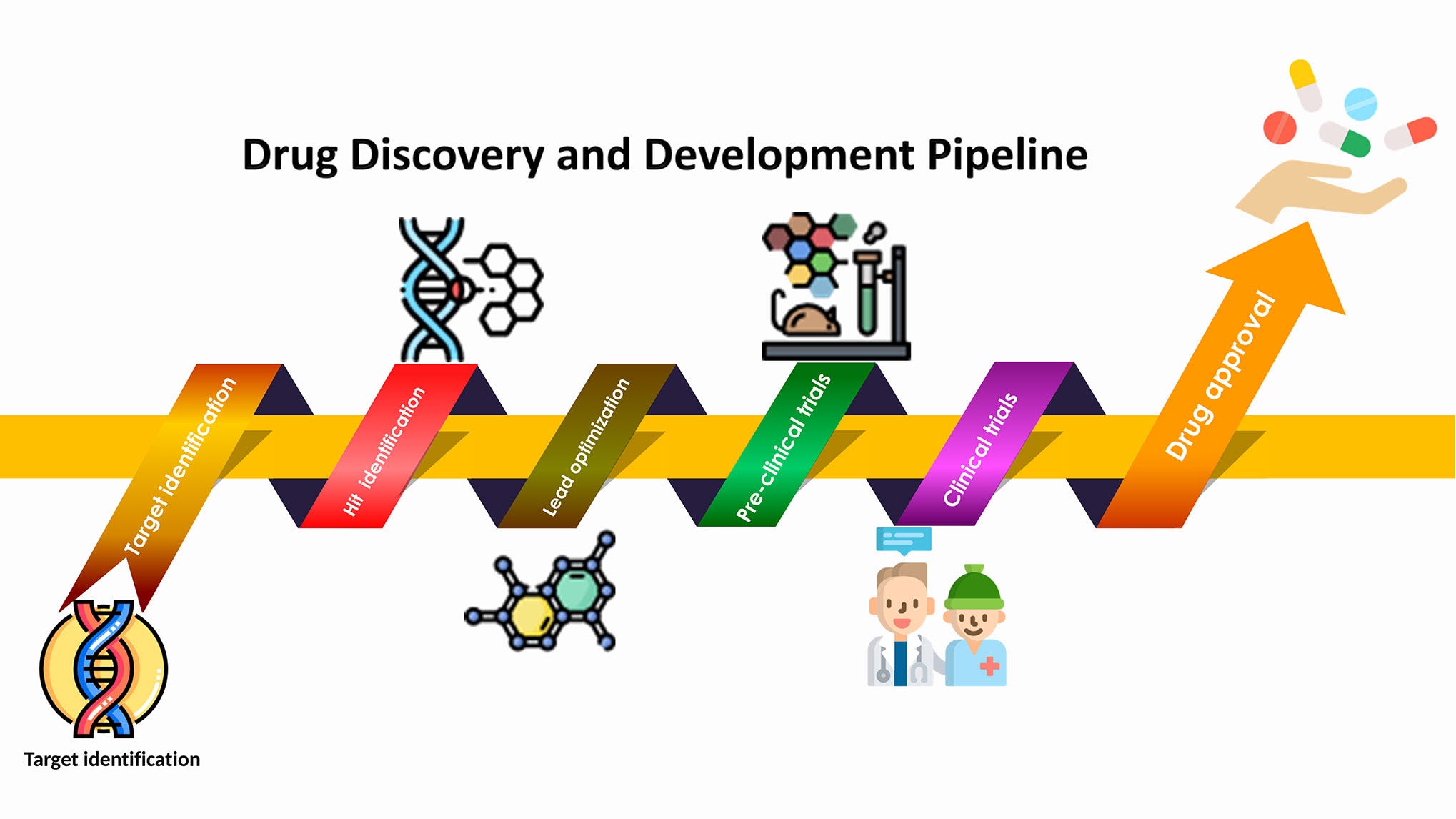
Computational methods are crucial throughout the process of discovering new drugs. The stages of drug discovery include:
Target Identification: Computational methods are utilized to identify potential biological targets based on molecular interactions, structural data, and disease pathways.
Target Validation: Tools are employed to validate identified targets by predicting their biological activity and assessing their relevance to disease processes.
'Hit' Identification: Virtual screening and molecular docking simulations are employed to identify potential 'hit' molecules that interact with the target of interest.
Lead Discovery: Computational models and simulations prioritize and predict the efficacy of 'hit' molecules in cell-based assays and animal models.
Hit-to-Lead (H2L): Computational assistance evaluates and selects lead candidates by predicting pharmacokinetic properties, metabolic stability, and other essential parameters.
Lead Optimization: Computational methods optimize lead molecules by predicting and modifying their chemical structures to enhance potency, selectivity, and safety profiles.
Preclinical Testing: During preclinical testing, scientists evaluate the safety and effectiveness of refined lead compounds using laboratory experiments and animal models. The results from these studies are then submitted to regulatory agencies to seek approval for advancing to clinical trials.
Clinical development involves several stages of testing in human subjects:
Phase 1: Initial safety testing in a small group of healthy volunteers.
Phase 2: Testing in a larger group of patients to assess effectiveness and further evaluate safety.
Phase 3: Large-scale testing in patients to confirm effectiveness, monitor side effects, and compare with existing treatments.
Key Tools In Computational Drug Design And Discovery
Molecular docking: It helps us understand how a drug molecule fits into its target protein, like finding the perfect key for a lock. This process estimates how strongly the drug and its target interact, which is essential in designing new drugs. It uses advanced tools like AutoDock Vina, AutoDock GOLD, Glide, DOCK, LigandFit, and SwissDock to get these predictions.
Pharmacophore modeling: identifies the key features in a molecule that are essential for its activity. This method is a cornerstone of modern drug discovery, helping scientists design new compounds with better pharmacological properties. It’s a powerful tool for medicinal chemists, making the creation of effective new drugs more precise and rational. Examples include LigandScout, Discovery Studio.
Quantitative Structure-Activity Relationship (QSAR) modeling: examines how the chemical structure of molecules relates to their biological activities. By using statistical methods, QSAR models can predict how new compounds will behave based on their structure. This helps chemists make smart changes to improve a drug's effectiveness or reduce its side effects. Examples include Software like Schrödinger's QSAR module, ADMET Predictor.
Challenges Of Computational Drug Design And Discovery
Although CADD offers advantages in speeding up drug discovery, it also comes with its own set of challenges such as:
Data Quality and Quantity: CADD predictions are only as good as the data they're based on. If the data is poor quality or insufficient, the predictions won't be reliable. High-quality, well-curated datasets are crucial, especially for machine learning in drug discovery. Cleaning up data by removing outliers and ensuring consistent formatting can help make these predictions more accurate.
Accuracy of Predictive Models: One major challenge in CADD is making sure our computational models are accurate. Since these models rely on theoretical frameworks, like molecular dynamics simulations and docking scores, they might not capture all the complexities of biological systems. To improve accuracy, we need to really understand the details of scoring algorithms.
Time and Computational Cost: Advanced CADD techniques, such as detailed molecular dynamics simulations and complex machine learning models, require significant computational power. This can be costly in terms of both time and infrastructure, which might be a barrier for some research groups.
Over-reliance on Computational Predictions: While CADD is a powerful tool, relying too much on its predictions without experimental validation can lead to wasted efforts. It's important to balance computational predictions with experimental evidence to ensure successful drug discovery.
Conclusion
Computer-Aided Drug Design (CADD) is revolutionizing modern drug discovery combining biology and computational power. As CADD evolves, it brings innovation, faces challenges, and addresses ethical considerations, aiming to make drug discovery faster, more accurate, and focused on patient well-being, leading to a healthier future for all.
References
Wenqiang Cui, Adnane Aouidate Yanhua Li et al(2020) Discovering anti cancer drugs via computational methods. https://doi.org/10.3389/fphar.2020.00733
Sarfaraz K. Niazi and Zamara Mariam (2023); Computer Aided Drug Design and Drug Discovery. 10.3390/ph17010022.
This is awesome.
This is a very nice article, Miss Hassan.
I have learnt how computation, modelling and stimulation helps in the discovery of new drugs.
Thanks for sharing